Gene therapy is the insertion of genes into an individual's cells and tissues to treat a disease, and hereditary diseases in which a defective mutant allele is replaced with a functional one. Although the technology is still in its infancy, it has been used with some success. Antisense therapy is not strictly a form of gene therapy, but is a genetically-mediated therapy and is often considered together with other methods.

Gene therapy using an Adenovirus vector. A new gene is inserted into an adenovirus vector, which is used to introduce the modified DNA into a human cell. If the treatment is successful, the new gene will make a functional protein.
Background
On September 14, 1990 at the U.S. National Institutes of Health W. French Anderson, M.D., and his colleagues R. Michael Blaese, M.D., C. Bouzaid, M.D., and Kenneth Culver, M.D., performed the first approved gene therapy procedure on four-year old Ashanthi DeSilva. Born with a rare genetic disease called severe combined immunodeficiency (SCID), she lacked a healthy immune system, and was vulnerable to every passing germ or infection. Children with this illness usually develop overwhelming infections and rarely survive to adulthood; a common childhood illness like chickenpox is life-threatening. Ashanthi led a cloistered existence -- avoiding contact with people outside her family, remaining in the sterile environment of her home, and battling frequent illnesses with massive amounts of antibiotics.
In Ashanthi's gene therapy procedure, doctors removed white blood cells from the child's body, let the cells grow in the lab, inserted the missing gene into the cells, and then infused the genetically modified blood cells back into the patient's bloodstream. Laboratory tests have shown that the therapy strengthened Ashanthi's immune system by 40%; she no longer has recurrent colds, she has been allowed to attend school, and she was immunized against whooping cough. This procedure was not a cure; the white blood cells treated genetically only work for a few months, after which the process must be repeated (VII, Thompson [First] 1993). As of early 2007, she was still in good health, and she was attending college. However, there is no consensus on what portion of her improvement should be attributed to gene therapy versus other treatments. Some would state that the case is of great importance despite its indefinite results, if only because it demonstrated that gene therapy could be practically attempted without adverse consequences.
Although this simplified explanation of a gene therapy procedure sounds like a happy ending, it is little more than an optimistic first chapter in a long story; the road to the first approved gene therapy procedure was rocky and fraught with controversy. The biology of human gene therapy is very complex, and there are many techniques that still need to be developed and diseases that need to be understood more fully before gene therapy can be used appropriately. The public policy debate surrounding the possible use of genetically engineered material in human subjects has been equally complex. Major participants in the debate have come from the fields of biology, government, law, medicine, philosophy, politics, and religion, each bringing different views to the discussion.
Scientists took the logical step of trying to introduce genes straight into human cells, focusing on diseases caused by single-gene defects, such as cystic fibrosis, hemophilia, muscular dystrophy and sickle cell anemia. However, this has been much harder than modifying simple bacteria, primarily because of the problems involved in carrying large sections of DNA and delivering them to the correct site on the comparatively large human genome.
Basic process
In most gene therapy studies, a "correct copy" or "wild type" gene is provided or inserted into the genome. Generally, it is not an exact replacement of the "abnormal," disease-causing gene, but rather extra, correct copies of genes are provided to complement the loss of function. A carrier called a vector must be used to deliver the therapeutic gene to the patient's target cells. Currently, the most common type of vectors are viruses that have been genetically altered to carry normal human DNA. Viruses have evolved a way of encapsulating and delivering their genes to human cells in a pathogenic manner. Scientists have tried to harness this ability by manipulating the viral genome to remove disease-causing genes and insert therapeutic ones.
Target cells such as the patient's liver or lung cells are infected with the vector. The vector then unloads its genetic material containing the therapeutic human gene into the target cell. The generation of a functional protein product from the therapeutic gene restores the target cell to a normal cell.
Types of gene therapy
Gene therapy may be classified into the following types:
Germ line gene therapy
In the case of germ line gene therapy, germ cells, i.e., sperm or eggs, are modified by the introduction of functional genes, which are ordinarily integrated into their genomes. Therefore, the change due to therapy would be heritable and would be passed on to later generations. This new approach, theoretically, should be highly effective in counteracting genetic disorders. However, this option is prohibited for application in human beings, at least for the present, for a variety of technical and ethical reasons.
Somatic gene therapy
In the case of somatic gene therapy, theraputic genes are transferred into the somatic cells of a patient. Any modifications and effects will be restricted to the individual patient only, and will not be inherited by the patient's offspring.
Broad methods
There are a variety of different methods to replace or repair the genes targeted in gene therapy.
- A normal gene may be inserted into a nonspecific location within the genome to replace a nonfunctional gene. This approach is most common.
- An abnormal gene could be swapped for a normal gene through homologous recombination.
- The abnormal gene could be repaired through selective reverse mutation, which returns the gene to its normal function.
- The regulation (the degree to which a gene is turned on or off) of a particular gene could be altered.
Vectors in gene therapy
Viruses
All viruses bind to their hosts and introduce their genetic material into the host cell as part of their replication cycle. This genetic material contains basic 'instructions' of how to produce more copies of these viruses, hijacking the body's normal production machinery to serve the needs of the virus. The host cell will carry out these instructions and produce additional copies of the virus, leading to more and more cells becoming infected. Some types of viruses physically insert their genes into the host's genome (a defining feature of retroviruses, the family of viruses that includes HIV, is that the virus will introduce the enzyme reverse transcriptase into the host and thus use its RNA as the "instructions"). This incorporates the genes of that virus among the genes of the host cell for the life span of that cell.
Doctors and molecular biologists realized that viruses like this could be used as vehicles to carry 'good' genes into a human cell. First, a scientist would remove the genes in the virus that cause disease. Then they would replace those genes with genes encoding the desired effect (for instance, insulin production in the case of diabetics). This procedure must be done in such a way that the genes which allow the virus to insert its genome into its host's genome are left intact. This can be confusing, and requires significant research and understanding of the virus' genes in order to know the function of each. An example: A virus is found which replicates by inserting its genes into the host cell's genome. This virus has two genes- A and B. Gene A encodes a protein which allows this virus to insert itself into the host's genome. Gene B causes the disease this virus is associated with. Gene C is the "normal" or "desirable" gene we want in the place of gene B. Thus, by re-engineering the virus so that gene B is replaced by gene C, while allowing gene A to properly function, this virus could introduce the required gene - gene C into the host cell's genome without causing any disease.
All this is clearly an oversimplification, and numerous problems exist that prevent gene therapy using viral vectors, such as: trouble preventing undesired effects, ensuring the virus will infect the correct target cell in the body, and ensuring that the inserted gene doesn't disrupt any vital genes already in the genome. However, this basic mode of gene introduction currently shows much promise and doctors and scientists are working hard to fix any potential problems that could exist.
Retroviruses
The genetic material in retroviruses is in the form of RNA molecules, while the genetic material of their hosts is in the form of DNA. When a retrovirus infects a host cell, it will introduce its RNA together with some enzymes, namely reverse transcriptase and integrase, into the cell. This RNA molecule from the retrovirus must produce a DNA copy from its RNA molecule before it can be integrated into the genetic material of the host cell. The process of producing a DNA copy from an RNA molecule is termed reverse transcription. It is carried out by one of the enzymes carried in the virus, called reverse transcriptase. After this DNA copy is produced and is free in the nucleus of the host cell, it must be incorporated into the genome of the host cell. That is, it must be inserted into the large DNA molecules in the cell (the chromosomes). This process is done by another enzyme carried in the virus called integrase.
Now that the genetic material of the virus is incorporated and has become part of the genetic material of the host cell, it can be said that the host cell is now modified to contain a new gene. If this host cell divides later, its descendants will all contain the new genes. Sometimes the genes of the retrovirus do not express their information immediately.
One of the problems of gene therapy using retroviruses is that the integrase enzyme can insert the genetic material of the virus in any arbitrary position in the genome of the host- it randomly shoves the genetic material into a chromosome. If genetic material happens to be inserted in the middle of one of the original genes of the host cell, this gene will be disrupted (insertional mutagenesis). If the gene happens to be one regulating cell division, uncontrolled cell division (i.e., cancer) can occur. This problem has recently begun to be addressed by utilizing zinc finger nucleases or by including certain sequences such as the beta-globin locus control region to direct the site of integration to specific chromosomal sites.
Gene therapy trials using retroviral vectors to treat X-linked severe combined immunodeficiency (X-SCID) represent the most successful application of gene therapy to date. More than twenty patients have been treated in France and Britain, with a high rate of immune system reconstitution observed. Similar trials were halted or restricted in the USA when leukemia was reported in patients treated in the French X-SCID gene therapy trial. To date, four children in the French trial and one in the British trial have developed leukemia as a result of insertional mutagenesis by the retroviral vector. All but one of these children responded well to conventional anti-leukemia treatment. Gene therapy trials to treat SCID due to deficiency of the Adenosine Deaminase (ADA) enzyme continue with relative success in the USA, Britain, Italy and Japan.
Adenoviruses
Adenoviruses are viruses that carry their genetic material in the form of double-stranded DNA. They cause respiratory, intestinal, and eye infections in humans (especially the common cold). When these viruses infect a host cell, they introduce their DNA molecule into the host. The genetic material of the adenoviruses is not incorporated (transient) into the host cell's genetic material. The DNA molecule is left free in the nucleus of the host cell, and the instructions in this extra DNA molecule are transcribed just like any other gene. The only difference is that these extra genes are not replicated when the cell is about to undergo cell division so the descendants of that cell will not have the extra gene. As a result, treatment with the adenovirus will require readministration in a growing cell population although the absence of integration into the host cell's genome should prevent the type of cancer seen in the SCID trials. This vector system has shown real promise in treating cancer and indeed the first gene therapy product to be licensed to treat cancer is an adenovirus.
Adeno-associated viruses
Adeno-associated viruses, from the parvovirus family, are small viruses with a genome of single stranded DNA. The wild type AAV can insert genetic material at a specific site on chromosome 19 with near 100% certainty. But the recombinant AAV, which does not contain any viral genes and only the therapeutic gene, does not integrate into the genome. Instead the recombinant viral genome fuses at its ends via the ITR (inverted terminal repeats) recombination to form circular, episomal forms which are predicted to be the primary cause of the long term gene expression. There are a few disadvantages to using AAV, including the small amount of DNA it can carry (low capacity) and the difficulty in producing it. This type of virus is being used, however, because it is non-pathogenic (most people carry this harmless virus). In contrast to adenoviruses, most people treated with AAV will not build an immune response to remove the virus and the cells that have been successfully treated with it. Several trials with AAV are on-going or in preparation, mainly trying to treat muscle and eye diseases; the two tissues where the virus seems particularly useful. However, clinical trials have also been initiated where AAV vectors are used to deliver genes to the brain. This is possible because AAV viruses can infect non-dividing (quiescent) cells, such as neurons in which their genomes are expressed for a long time.
Envelope protein pseudotyping of viral vectors
The viral vectors described above have natural host cell populations that they infect most efficiently. Retroviruses have limited natural host cell ranges, and although adenovirus and adeno-associated virus are able to infect a relatively broader range of cells efficiently, some cell types are refractory to infection by these viruses as well. Attachment to and entry into a susceptible cell is mediated by the protein envelope on the surface of a virus. Retroviruses and adeno-associated viruses have a single protein coating their membrane, while adenoviruses are coated with both an envelope protein and fibers that extend away from the surface of the virus. The envelope proteins on each of these viruses bind to cell-surface molecules such as heparin sulfate, which localizes them upon the surface of the potential host, as well as with the specific protein receptor that either induces entry-promoting structural changes in the viral protein, or localizes the virus in endosomes wherein acidification of the lumen induces this refolding of the viral coat. In either case, entry into potential host cells requires a favorable interaction between a protein on the surface of the virus and a protein on the surface of the cell. For the purposes of gene therapy, one might either want to limit or expand the range of cells susceptible to transduction by a gene therapy vector. To this end, many vectors have been developed in which the endogenous viral envelope proteins have been replaced by either envelope proteins from other viruses, or by chimeric proteins. Such chimera would consist of those parts of the viral protein necessary for incorporation into the virion as well as sequences meant to interact with specific host cell proteins. Viruses in which the envelope proteins have been replaced as described are referred to as pseudotyped viruses. For example, the most popular retroviral vector for use in gene therapy trials has been the lentivirus Simian immunodeficiency virus coated with the envelope proteins, G-protein, from Vesicular stomatitis virus. This vector is referred to as VSV G-pseudotyped lentivirus, and infects an almost universal set of cells. This tropism is characteristic of the VSV G-protein with which this vector is coated. Many attempts have been made to limit the tropism of viral vectors to one or a few host cell populations. This advance would allow for the systemic administration of a relatively small amount of vector. The potential for off-target cell modification would be limited, and many concerns from the medical community would be alleviated. Most attempts to limit tropism have used chimeric envelope proteins bearing antibody fragments. These vectors show great promise for the development of "magic bullet" gene therapies.
Non-viral methods
Non-viral methods present certain advantages over viral methods, with simple large scale production and low host immunogenicity being just two. Previously, low levels of transfection and expression of the gene held non-viral methods at a disadvantage; however, recent advances in vector technology have yielded molecules and techniques with transfection efficiencies similar to those of viruses.
Naked DNA
This is the simplest method of non-viral transfection. Clinical trials carried out of intramuscular injection of a naked DNA plasmid have occurred with some success; however, the expression has been very low in comparison to other methods of transfection. In addition to trials with plasmids, there have been trials with naked PCR product, which have had similar or greater success. This success, however, does not compare to that of the other methods, leading to research into more efficient methods for delivery of the naked DNA such as electroporation, sonoporation, and the use of a "gene gun", which shoots DNA coated gold particles into the cell using high pressure gas.
Oligonucleotides
The use of synthetic oligonucleotides in gene therapy is to inactivate the genes involved in the disease process. There are several methods by which this is achieved. One strategy uses antisense specific to the target gene to disrupt the transcription of the faulty gene. Another uses small molecules of RNA called siRNA to signal the cell to cleave specific unique sequences in the mRNA transcript of the faulty gene, disrupting translation of the faulty mRNA, and therefore expression of the gene. A further strategy uses double stranded oligodeoxynucleotides as a decoy for the transcription factors that are required to activate the transcription of the target gene. The transcription factors bind to the decoys instead of the promoter of the faulty gene, which reduces the transcription of the target gene, lowering expression. Additionally, single stranded DNA oligonucleotides have been used to direct a single base change within a mutant gene. The oligonucleotide is designed to anneal with complementarity to the target gene with the exception of a central base, the target base, which serves as the template base for repair. This technique is referred to as oligonucleotide mediated gene repair, targeted gene repair, or targeted nucleotide alteration.
Lipoplexes and polyplexes
To improve the delivery of the new DNA into the cell, the DNA must be protected from damage and its entry into the cell must be facilitated. To this end new molecules, lipoplexes and polyplexes, have been created that have the ability to protect the DNA from undesirable degradation during the transfection process.
Plasmid DNA can be covered with lipids in an organized structure like a micelle or a liposome. When the organized structure is complexed with DNA it is called a lipoplex. There are three types of lipids, anionic (negatively charged), neutral, or cationic (positively charged). Initially, anionic and neutral lipids were used for the construction of lipoplexes for synthetic vectors. However, in spite of the facts that there is little toxicity associated with them, that they are compatible with body fluids and that there was a possibility of adapting them to be tissue specific; they are complicated and time consuming to produce so attention was turned to the cationic versions.
Cationic lipids, due to their positive charge, were first used to condense negatively charged DNA molecules so as to facilitate the encapsulation of DNA into liposomes. Later it was found that the use of cationic lipids significantly enhanced the stability of lipoplexes. Also as a result of their charge, cationic liposomes interact with the cell membrane, endocytosis was widely believed as the major route by which cells uptake lipoplexes. Endosomes are formed as the results of endocytosis, however, if genes can not be released into cytoplasm by breaking the membrane of endosome, they will be sent to lysosomes where all DNA will be destroyed before they could achieve their functions. It was also found that although cationic lipids themselves could condense and encapsulate DNA into liposomes, the transfection efficiency is very low due to the lack of ability in terms of “endosomal escaping”. However, when helper lipids (usually electroneutral lipids, such as DOPE) were added to form lipoplexes, much higher transfection efficiency was observed. Later on, it was figured out that certain lipids have the ability to destabilize endosomal membranes so as to facilitate the escape of DNA from endosome, therefore those lipids are called fusogenic lipids. Although cationic liposomes have been widely used as an alternative for gene delivery vectors, a dose dependent toxicity of cationic lipids were also observed which could limit their therapeutic usages.
The most common use of lipoplexes has been in gene transfer into cancer cells, where the supplied genes have activated tumor suppressor control genes in the cell and decrease the activity of oncogenes. Recent studies have shown lipoplexes to be useful in transfecting respiratory epithelial cells, so they may be used for treatment of genetic respiratory diseases such as cystic fibrosis.
Complexes of polymers with DNA are called polyplexes. Most polyplexes consist of cationic polymers and their production is regulated by ionic interactions. One large difference between the methods of action of polyplexes and lipoplexes is that polyplexes cannot release their DNA load into the cytoplasm, so to this end, co-transfection with endosome-lytic agents (to lyse the endosome that is made during endocytosis, the process by which the polyplex enters the cell) such as inactivated adenovirus must occur. However, this isn't always the case, polymers such as polyethylenimine have their own method of endosome disruption as does chitosan and trimethylchitosan.
Hybrid methods
Due to every method of gene transfer having shortcomings, there have been some hybrid methods developed that combine two or more techniques. Virosomes are one example; they combine liposomes with an inactivated HIV or influenza virus. This has been shown to have more efficient gene transfer in respiratory epithelial cells than either viral or liposomal methods alone. Other methods involve mixing other viral vectors with cationic lipids or hybridising viruses.
Dendrimers
A dendrimer is a highly branched macromolecule with a spherical shape. The surface of the particle may be functionalized in many ways and many of the properties of the resulting construct are determined by its surface.
In particular it is possible to construct a cationic dendrimer, i.e. one with a positive surface charge. When in the presence of genetic material such as DNA or RNA, charge complimentarity leads to a temporary association of the nucleic acid with the cationic dendrimer. On reaching its destination the dendrimer-nucleic acid complex is then taken into the cell via endocytosis.
In recent years the benchmark for transfection agents has been cationic lipids. Limitations of these competing reagents have been reported to include: the lack of ability to transfect a number of cell types, the lack of robust active targeting capabilities, incompatibility with animal models, and toxicity. Dendrimers offer robust covalent construction and extreme control over molecule structure, and therefore size. Together these give compelling advantages compared to existing approaches.
Producing dendrimers has historically been a slow and expensive process consisting of numerous slow reactions, an obstacle that severely curtailed their commercial development. The Michigan based company Dendritic Nanotechnologies discovered a method to produce dendrimers using kinetically driven chemistry, a process that not only reduced cost by a magnitude of three, but also cut reaction time from over a month to several days. These new "Priostar" dendrimers can be specifically constructed to carry a DNA or RNA payload that transfects cells at a high efficiency with little or no toxicity.
Major developments in gene therapy
2002 and earlier
New gene therapy approach repairs errors in messenger RNA derived from defective genes. This technique has the potential to treat the blood disorder thalassaemia, cystic fibrosis, and some cancers. See Subtle gene therapy tackles blood disorder at NewScientist.com (October 11, 2002).
Researchers at Case Western Reserve University and Copernicus Therapeutics are able to create tiny liposomes 25 nanometers across that can carry therapeutic DNA through pores in the nuclear membrane. See DNA nanoballs boost gene therapy at NewScientist.com (May 12, 2002).
Sickle cell disease is successfully treated in mice. See Murine Gene Therapy Corrects Symptoms of Sickle Cell Disease from March 18, 2002, issue of The Scientist.
The success of a multi-center trial for treating children with SCID (severe combined immune deficiency or "bubble boy" disease) held from 2000 and 2002 was questioned when two of the ten children treated at the trial's Paris center developed a leukemia-like condition. Clinical trials were halted temporarily in 2002, but resumed after regulatory review of the protocol in the United States, the United Kingdom, France, Italy, and Germany. (V. Cavazzana-Calvo, Thrasher and Mavilio 2004; see also 'Miracle' gene therapy trial halted at NewScientist.com, October 3, 2002). NO current resourse aquired
In 1993 Andrew Gobea was born with a rare, normally fatal genetic disease - severe combined immunodeficiency (SCID). Genetic screening before birth showed that he had SCID. Blood was removed from Andrew's placenta and umbilical cord immediately after birth, containing stem cells. The allele that codes for ADA was obtained and was inserted into a retrovirus. Retroviruses and stem cells were mixed, after which they entered and inserted the gene into the stem cells' chromosomes. Stem cells containing the working ADA gene were injected into Andrew's blood system via a vein. For four years T-cells (white blood cells), produced by stem cells, made ADA enzymes using the ADA gene. After four years more treatment was needed.
2003
In 2003 a University of California, Los Angeles research team inserted genes into the brain using liposomes coated in a polymer called polyethylene glycol (PEG). The transfer of genes into the brain is a significant achievement because viral vectors are too big to get across the "blood-brain barrier." This method has potential for treating Parkinson's disease. See Undercover genes slip into the brain at NewScientist.com (March 20, 2003).
RNA interference or gene silencing may be a new way to treat Huntington's. Short pieces of double-stranded RNA (short, interfering RNAs or siRNAs) are used by cells to degrade RNA of a particular sequence. If a siRNA is designed to match the RNA copied from a faulty gene, then the abnormal protein product of that gene will not be produced. See Gene therapy may switch off Huntington's at NewScientist.com (March 13, 2003).
2006
Scientists at the National Institutes of Health (Bethesda, Maryland) have successfully treated metastatic melanoma in two patients using killer T cells genetically retargeted to attack the cancer cells. This study constitutes the first demonstration that gene therapy can be effective in treating cancer. The study results have been published in Science (October 2006).
In May 2006 a team of scientists led by Dr. Luigi Naldini and Dr. Brian Brown from the San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET) in Milan, Italy reported a breakthrough for gene therapy in which they developed a way to prevent the immune system from rejecting a newly delivered gene. Similar to organ transplantation, gene therapy has been plagued by the problem of immune rejection. So far, delivery of the 'normal' gene has been difficult because the immune system recognizes the new gene as foreign and rejects the cells carrying it. To overcome this problem, the HSR-TIGET group utilized a newly uncovered network of genes regulated by molecules known as microRNAs. Dr. Naldini's group reasoned that they could use this natural function of microRNA to selectively turn off the identity of their therapeutic gene in cells of the immune system and prevent the gene from being found and destroyed. The researchers injected mice with the gene containing an immune-cell microRNA target sequence, and spectacularly, the mice did not reject the gene, as previously occurred when vectors without the microRNA target sequence were used. This work will have important implications for the treatment of hemophilia and other genetic diseases by gene therapy.
In March 2006 an international group of scientists announced the successful use of gene therapy to treat two adult patients for a disease affecting myeloid cells. The study, published in Nature Medicine, is believed to be the first to show that gene therapy can cure diseases of the myeloid system.
2007
On 1 May 2007 Moorfields Eye Hospital and University College London's Institute of Ophthalmology announced the world's first gene therapy trial for inherited retinal disease. The first operation was carried out on a 23 year-old British male, Robert Johnson, in early 2007. Leber's congenital amaurosis is an inherited blinding disease caused by mutations in the RPE65 gene. The results of the Moorfields/UCL trial were published in New England Journal of Medicine in April 2008. They researched the safety of the subretinal delivery of recombinant adeno associated virus (AAV) carrying RPE65 gene, and found it yielded positive results, with patients having modest increase in vision, and, perhaps more importantly, no apparent side-effects.
Problems and ethics
For the safety of gene therapy, the Weismann barrier is fundamental in the current thinking. Soma-to-germline feedback should therefore be impossible. However, there are indications that the Weissman barrier can be breached. One way it might possibly be breached is if the treatment were somehow misapplied and spread to the testes and therefore would infect the germline against the intentions of the therapy.
Some of the problems of gene therapy include:
- Short-lived nature of gene therapy - Before gene therapy can become a permanent cure for any condition, the therapeutic DNA introduced into target cells must remain functional and the cells containing the therapeutic DNA must be long-lived and stable. Problems with integrating therapeutic DNA into the genome and the rapidly dividing nature of many cells prevent gene therapy from achieving any long-term benefits. Patients will have to undergo multiple rounds of gene therapy.
- Immune response - Anytime a foreign object is introduced into human tissues, the immune system has evolved to attack the invader. The risk of stimulating the immune system in a way that reduces gene therapy effectiveness is always a possibility. Furthermore, the immune system's enhanced response to invaders it has seen before makes it difficult for gene therapy to be repeated in patients.
- Problems with viral vectors - Viruses, while the carrier of choice in most gene therapy studies, present a variety of potential problems to the patient --toxicity, immune and inflammatory responses, and gene control and targeting issues. In addition, there is always the fear that the viral vector, once inside the patient, may recover its ability to cause disease.
- Multigene disorders - Conditions or disorders that arise from mutations in a single gene are the best candidates for gene therapy. Unfortunately, some of the most commonly occurring disorders, such as heart disease, high blood pressure, Alzheimer's disease, arthritis, and diabetes, are caused by the combined effects of variations in many genes. Multigene or multifactorial disorders such as these would be especially difficult to treat effectively using gene therapy.
- Chance of inducing a tumor (insertional mutagenesis) - If the DNA is integrated in the wrong place in the genome, for example in a tumor suppressor gene, it could induce a tumor. This has occurred in clinical trials for X-linked severe combined immunodeficiency (X-SCID) patients, in which hematopoietic stem cells were transduced with a corrective transgene using a retrovirus, and this led to the development of T cell leukemia in 3 of 20 patients.
- Religious concerns - Religious groups and creationists may consider the alteration of an individual's genes as tampering or corrupting God's work.
Deaths have occurred due to gene therapy, including that of Jesse Gelsinger.
In popular culture
- In the TV series Dark Angel gene therapy is mentioned as one of the practices performed on transgenics and their surrogate mothers at Manticore, and in the episode Prodigy, Dr. Tanaka uses a groundbreaking new form of gene therapy to turn Jude, a premature, vegitative baby of a crack/cocaine addict, into a boy genius.
- Gene therapy is a crucial plot element in the video game Metal Gear Solid, where it has been used to enhance the battle capabilities of enemy soldiers.
- Gene therapy plays a major role in the sci-fi series Stargate Atlantis, as a certain type of alien technology can only be used if one has a certain gene which is given to the members of the team through gene therapy.
- Gene therapy also plays a major role in the plot of the James Bond movie Die Another Day.
- The Yellow Bastard from Frank Miller's Sin City was also apparently the recipient of gene therapy.
- In the The Dark Knight Strikes Again, Dick Grayson, the first Robin, becomes a victim of extensive gene therapy for years by Lex Luthor to become The Joker.
- Gene therapy plays a recurring role in the present-time sci-fi television program ReGenesis, where it is used to cure various diseases, enhance athletic performance and produce vast profits for bio-tech corporations. (e.g. an undetectable performance-enhancing gene therapy was used by one of the characters on himself, but to avoid copyright infringement, this gene therapy was modified from the tested-to-be-harmless original, which produced a fatal cardiovascular defect)
- Gene therapy is the basis for the plot line of the film I Am Legend.
- Gene therapy is an important plot key in the game Bioshock where the game contents refer to plasmids and [gene] splicers.
- The book Next by Michael Crichton unravels a story in which fictitious biotechnology companies which experiment with gene therapy are involved.
- In the television show Alias, a breakthrough in molecular gene therapy is discovered, whereby a patient's body is reshaped to identically resemble someone else. Protagonist Sydney Bristow's best friend was secretly killed and her "double" resumed her place.






 .
.




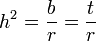 where r can be thought of as the coefficient of relatedness, b is the coefficient of regression and t the coefficient of correlation.
where r can be thought of as the coefficient of relatedness, b is the coefficient of regression and t the coefficient of correlation.


 ,
,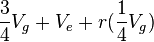

 term is the intraclass correlation among half sibs. We can easily calculate
term is the intraclass correlation among half sibs. We can easily calculate  . The Expected Mean Square is calculated from the relationship of the individuals (progeny within a sire are all half-sibs, for example), and an understanding of intraclass correlations.
. The Expected Mean Square is calculated from the relationship of the individuals (progeny within a sire are all half-sibs, for example), and an understanding of intraclass correlations.








